Let It Grow: FDA Revises Q&A Guidance to Support Sponsors With the Implementation of Expanded Access Programs
| Footnotes for this article are available at the end of this page. |
“Let it grow, let it blossom,” said Eric Clapton, and so the Food and Drug Administration expects of expanded access programs with the issuance of its guidance on “Expanded Access to Investigational Drugs for Treatment Use: Questions and Answers” (October 22, 2025). The guidance provides sponsors and providers with clearer direction on regulatory requirements and best practices for implementing expanded access programs.
As background, in 2009, FDA revised its Investigational New Drug (“IND”) regulations to create three categories of expanded access:
- Individual patient (including emergency uses)
- Intermediate-size patient population
- Widespread treatment use through a treatment IND or protocol
In response to the 2009 final rule FDA issued, a 2017 guidance document aimed to help industry implement the regulatory requirements for expanded access. Since 2017, FDA received additional questions and comments, particularly concerning statutory requirements from the 21st Century Cures Act and the FDA Reauthorization Act of 2017. The 2025 guidance is an attempt to address some of these concerns. FDA expects public availability of this guidance to help industry understand expanded access requirements and inform patients and healthcare providers on how to obtain investigational drugs when no approved treatments exist.
In this Bulletin, we will highlight some key points of the guidance and offer our own observations.
Highlights
- The guidance reassures the public that multiple safeguards are in place for expanded access use: safety reporting, physician training and oversight, informed consent, and recordkeeping requirements, to name a few.
- The guidance addresses questions raised by industry in response to two key statutory changes:
- The 21st Century Cures Act added section 561A to the Federal Food, Drug, and Cosmetic Act (“FD&C Act”), requiring manufacturers to make their expanded access policies publicly available.1
- The FDA Reauthorization Act of 2017 (“FDARA”) amended the FD&C Act to require that a drug’s expanded access policy be posted by either the start of a phase 2/3 trial or within 15 days of receiving fast track, breakthrough, or regenerative advanced therapy designation. Posting the policy does not guarantee access to the product, which remains voluntary.2
- The guidance outlines the regulatory pathways and key requirements for submitting expanded access requests.
- There are two regulatory pathways to expanded access: (1) an expanded access protocol to amend an existing IND; or (2) a new IND submission. An expanded access protocol should only be used when a sponsor seeking expanded access already has an existing IND in effect.
- FDA categorizes all expanded access requests based on the regulatory pathway and category of expanded access:
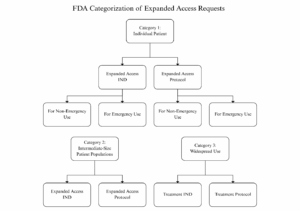
-
- Appendix A to the guidance summarizes the various expanded access request types, including required waiting periods and IRB requirements.
- For each subcategory of expanded access request, FDA clarifies what forms and information are required, including whether IRB review is required and who can initiate the request (the IND sponsor or licensed physician).
- Emergency use requests are only appropriate when treatment must occur within a matter of hours.3
- FDA clarifies that all expanded access requests are subject to informed consent requirements.
- For ease of use, the agency shares an informed consent template in Appendix B for individual patient expanded access. The template contains model language and sample signature/contact sections.
- Additional safeguards for children continue to apply. Those can be found in 21 C.F.R. Part 50, Subpart D.
- The guidance reviews the criteria that physicians and FDA must use in determining the appropriateness of expanded use for individual patients.
- The patient’s physician must determine whether the “probable risk” to the patient from the investigational drug is not greater than the probable risk from the disease or condition.
- The agency determines whether the potential benefit justifies the potential risks of the treatment use with the drug.
- The agency also determines whether the patient has a “serious or life-threatening condition” and has no other “comparable or satisfactory therapeutic options.
- For intermediate-size patient populations (which can be more than one person) and widespread use, sponsors can consolidate expanded access in a single IND or protocol.
- Multiple INDs or protocols for the same drug and condition are allowed but expected to be rare.
- Any number of patients beyond one could be appropriate for intermediate-size patient population protocol.
- When the number of patients continues to grow, sponsors should consult FDA to transition to a treatment IND or protocol. The agency itself may ask the sponsor to make the transition.
- Treatment can begin once the sponsor has agreed to provide the drug and all regulatory requirements are met:
- For emergency use INDs or protocols, treatment may start immediately upon FDA authorization, with a written request to follow within 15 working days, and the IRB must be notified within five working days.
- For non-emergency INDs, treatment may begin when the IND goes into effect, typically 30 days after FDA receipt and IRB approval.
- For non-emergency protocols, treatment may begin once the protocol has been submitted to FDA and IRB approval.
- For widespread use treatment protocols, there is also a 30-day waiting period unless FDA authorizes an earlier start.
- FDA authorizes expanded access only if it will not impede ongoing or planned clinical trials that could support marketing approval, generally limiting access to patients otherwise ineligible or unable to join such trials.
- Sponsors must follow all safety reporting rules for expanded access, including reporting serious unexpected reactions, submitting annual reports, and summarizing outcomes at the end of treatment. FDA reviews adverse event data to spot early safety signals but rarely stops INDs based on expanded access treatment results.
- To comply with the Cures Act, sponsors must publicly post their expanded access policies, including contact information, submission procedures, evaluation criteria, response timelines, and links to ClinicalTrials.gov.
- Policies posted should focus on providing clear, factual guidance about how patients and physicians can request access, the criteria for evaluating requests, and relevant regulatory references, without making any claims about the drug’s safety, effectiveness, or superiority.
- FDARA requires manufacturers or developers of investigational drugs for serious conditions to make their expanded access policies public by either the start of a phase 2/3 trial or within 15 days of receiving breakthrough, fast track, or regenerative advanced therapy designation.
AGG Observations
- Industry should leverage existing INDs and consolidate protocols whenever possible to streamline expanded access requests, facilitate IRB review, and minimize interference with ongoing or planned clinical trials.
- Sponsors should use Appendix B’s informed consent template for individual patient expanded access. It includes model language and sample signature/contact sections. This addresses a gap from the 2017 guidance and offers an opportunity for sponsors to streamline consent preparation, ensure regulatory compliance, and reduce administrative burden.
- FDA remains concerned that expanded access for large patient populations can risk slowing drug development, so sponsors should provide detailed plans demonstrating how trials and drug development will not be disrupted before seeking authorization.
- When submitting expanded access requests, sponsors should proactively provide comprehensive data and support to ensure FDA and treating physicians can confidently determine that the investigational drug’s potential benefits outweigh its risks, that the patient faces a serious or life-threatening condition, and that no comparable or satisfactory therapeutic alternatives exist.
- When publicly posting expanded access policies to comply with the Cures Act, sponsors should avoid language that could be interpreted as marketing or endorsing the drug for a particular use, and any references to the investigational drug should be neutral, factual, and limited to its availability under expanded access.
- Sponsors should post expanded access policies by phases 2 or 3 start or within 15 days of breakthrough, fast track, or regenerative advanced therapy designation, so physicians and patients have clear guidance.
- FDA hopes its newly issued guidance allows expanded access programs to grow. Companies that embrace the guidance and follow the agency’s recommendations may see their programs blossom.
[1] 21 U.S.C. 360bbb-0(b).
[2] 21 U.S.C. 360bbb-0(f).
[3] FDA does not define a specific number of hours but generally describes appropriate cases for emergency use requests as those requiring immediate treatment to prevent serious harm or death–situations where there is not enough time for a written submission.
- Alan G. Minsk
Partner
- Laura Dona LaBrie
Associate
- Aditya Krishnaswamy
Associate


